An automated framework for high-throughput predictions of NMR chemical shifts within liquid solutions
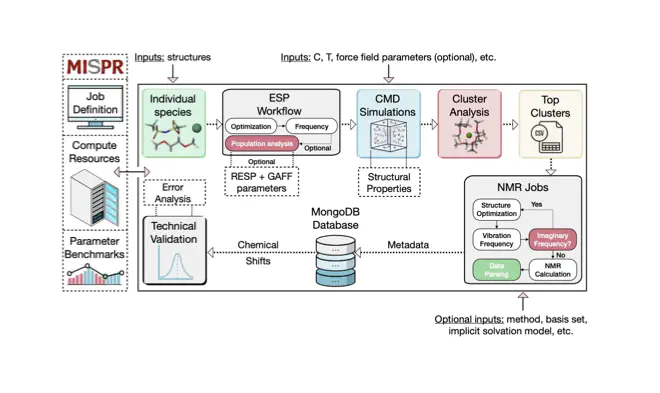 Overview of the NMR framework
Overview of the NMR frameworkAbstract
Identifying stable speciation in multi-component liquid solutions is fundamentally important to areas from electrochemistry to organic chemistry and biomolecular systems. Here we introduce a fully automated, high-throughput computational framework for the accurate prediction of stable species in liquid solutions by computing the nuclear magnetic resonance (NMR) chemical shifts. The framework automatically extracts and categorizes hundreds of thousands of atomic clusters from classical molecular dynamics simulations, identifies the most stable species in solution and calculates their NMR chemical shifts via density functional theory calculations. Additionally, the framework creates a database of computed chemical shifts for liquid solutions across a wide chemical and parameter space. We compare our computational results to experimental measurements for magnesium bis(trifluoromethanesulfonyl)imide Mg(TFSI)2 salt in dimethoxyethane solvent. Our analysis of the Mg2+ solvation structural evolutions reveals key factors that influence the accuracy of NMR chemical shift predictions in liquid solutions. Furthermore, we show how the framework reduces the performance of over 300 13C and 600 1H density functional theory chemical shift predictions to a single submission procedure.
Rasha Atwi
Scientist, Computational Chemistry
My research interests include high-throughput scientific computing, quantum chemistry, and molecular dynamics.